Boston MedTech Advisors Blog, January 2024
The Breakthrough Devices Program, established by the FDA in 2018, is a voluntary program for certain medical devices and device-led combination products that provide for more effective treatment or diagnosis of life-threatening or irreversibly debilitating diseases or conditions. It is available for devices which are subject to review under a premarket approval application (PMA), 510(k) application, or a De Novo submissions, alike. The vision of the program is to help patients gain timely access to certain necessary medical devices through expedited development and review while ensuring their safety and efficacy. This program replaced the Expedited Access Pathway (EAP) and Priority Review for medical devices programs.
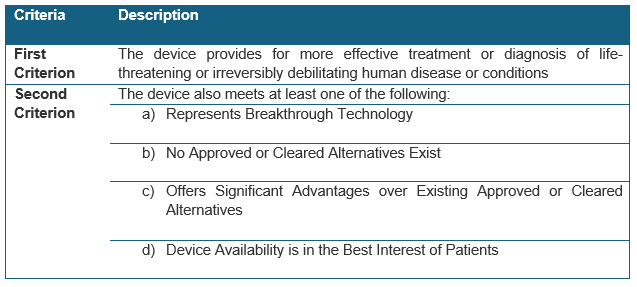
To be eligible for classification as a breakthrough device, a device must meet the first of the following criteria and at least one of the sub-sections in the second criterion:1,2
Breakthrough device designation (BDD) decisions are made prior to marketing authorization and complete clinical data demonstrating the device’s safety and effectiveness may not be available during the breakthrough request review process. Therefore, in its review, FDA considers whether the sponsor has demonstrated a ‘reasonable expectation’ of technical success (i.e., the device can be built and function as intended) as well as clinical success (i.e., the device could more effectively treat or diagnose the identified disease or condition).
In September 2023, FDA published its final guidance on the Breakthrough Devices Program. This guidance document, which supersedes FDA’s 2018 guidance on the Breakthrough Devices Program, provides specific insights into the agency’s current thinking on the types of devices that may be considered as breakthrough devices.
Manufacturers of medical devices and device-led combination products that meet the criteria can apply for BDD at any time before the submission of marketing authorization application and, if accepted, can expect benefits including:
- Interactive communication with FDA throughout the device development and review processes.
- Increased reliance on post-market data collection for PMAs designated as Breakthrough Devices for expedited development and market authorization.
- Efficient, adaptive, and flexible clinical study design with suitable endpoints such as intermediate and surrogate endpoints or composite endpoints.
- Priority review of their premarket application.
To facilitate patients’ access to breakthrough device technologies, FDA and the Centers for Medicare and Medicaid Services (CMS) have been working in tandem on a solution that may reduce the delay between FDA approval and reimbursement coverage by CMS. In January 2021 CMS published the “Medicare Coverage of Innovative Technology and Definition of ‘Reasonable and Necessary’” (MCIT/R&N) final rule intended to grant expedited Medicare coverage for up to four years for any FDA-designated breakthrough device that received market authorization. However, public comments raised concerns regarding the lack of risk-benefit information to Medicare beneficiaries. Therefore, this rule was repealed in November 2021 in favor of a coverage process that covers new technologies on the basis of scientifically sound clinical evidence, with appropriate health and safety protections in place for the Medicare population.
Consequently, in June 2023, CMS published a draft guideline for “Transitional Coverage for Emerging Technologies” (TCET). The new initiative is intended to provide expedited coverage for new technologies designated by the FDA as Breakthrough Devices, [see BMTA blog for more information]. Through this program, CMS will finalize National Coverage Determinations (NCDs) for technologies selected for the TCET pathway within 6 months of FDA market authorization (a process that typically takes an average of five years for novel medical devices) thereby significantly shortening the lag time between FDA approval and CMS coverage.
In September 2023, FDA published its final guidance on the Breakthrough Devices Program.1 This guidance document, which supersedes FDA’s 2018 guidance on the Breakthrough Devices Program,3 provides specific insights into the agency’s current thinking on the types of devices that may be considered as breakthrough devices.
While the 2023 guidance does not introduce any new changes in the designation criteria, it clarifies and expands the types of devices that may be eligible for the Breakthrough Devices Program compared to the 2018 guidance, as follows:
- Clarifies Criterion 1 designation considerations:
- While in the 2018 guidance document the mechanisms for demonstrating ‘reasonable expectation’ of technical and clinical success could include literature or preliminary data, including bench, animal, or clinical data, the current guidance clarifies that evidence may vary based on the intended use of the device, its technology and features, and the available standard of care alternatives (e.g., the regulatory bar may be higher if a treatment requesting BDD already has a long-standing and established standard of care).
- The guidance clarifies that FDA considers the ‘totality of information’, which includes the intended use, potential for technical and clinical success, potential for clinically meaningful impact, and potential benefits and risks.
- Expands the criteria for breakthrough devices to include products aiming at achieving health equity (fair opportunity to attain their highest level of health for everyone) and eliminate disparities in health care:
- Technologies capable of achieving health equity by addressing health outcomes in diverse populations (e.g., different race, ethnicity, sex, age, disability, sexual orientation, gender identity, socioeconomic status, geography, preferred language, etc.).
- Devices that have the potential to offer a clinically meaningful impact through improved accessibility and thus provide significant benefit to patients (i.e., ease of use by diverse populations or in more diverse settings).
- Products intended to diagnose or treat rare diseases.
- Expands the criteria for breakthrough devices to include products that offer non-addictive treatments for pain or addiction. This was done to fulfil FDAs obligations under the 2018 Substance Use-Disorder Prevention, that Promotes Opioid Recovery and Treatment for Patients and Communities Act (SUPPORT).
- Clarifies FDA’s policy regarding the disclosure of BDD requests and the agency’s decisions. Except when the designation request has been publicly acknowledged by the device sponsor, the agency will not disclose such information. However, FDA maintains and publishes a list of breakthrough devices that have been market authorized on their Breakthrough Devices Program webpage.2
As of the end of 2023, FDA has granted 921 BDDs.4 The top five clinical panels of these breakthrough designated devices, by order, were cardiovascular, neurology, orthopedic, gastroenterology/urology and general/plastic surgery. Of those BDD devices, 77 devices subsequently received marketing authorization by CDRH (24 PMAs, 26 510(k)s, and 27 De-Novo submissions).2 In 2023 alone, CDRH granted the BDD request to 167 devices and granted marketing authorization to 29 of them.2
Of note, devices that are not eligible for BDD because they are not intended for the treatment or diagnosis of a life-threatening or irreversibly debilitating human disease or condition, may be considered for the Safer Technologies Program (STeP).5 Candidates for this program include devices that are reasonably expected to significantly improve the benefit-risk profile of a treatment or diagnostic through substantial safety innovations (i.e., provide reduction in serious adverse event, device failure mode, use-related hazard or use error, or improvement in the safety of another device or intervention). The aim of the STeP program is similarly intended to help reduce the time it takes to develop and obtain marketing authorization. It provides manufacturers an opportunity to have facilitated and timely interactions with FDA’s experts throughout the premarket review phase. There are currently 35 devices included in the STeP program. In 2023, 15 STeP Devices were enrolled and two were authorized for marketing.4
In summary, device manufacturers should consider whether they may qualify for the Breakthrough Device program early in their development process and take full advantage of the benefits of working more closely with FDA during the development process. Manufacturers should keep abreast of FDA’s latest expansions to devices that may qualify for the program, as there are significant regulatory, marketing, and reimbursement implications of receiving such a designation.
About Boston MedTech Advisors (BMTA):
Since 2004, BMTA’s multidisciplinary team of highly experienced consultants has supported more than 400 medical technologies and life sciences companies around the world to achieve their business goals. BMTA assists its clients to commercialize new products and services and increase their market adoption, by addressing their unique and inter-dependent regulatory, clinical, reimbursement, marketing, and business development requirements. BMTA offers valuable, ethical, result-oriented, professional, and cost-effective insights that recognize the multi-faceted aspects of today’s healthcare markets and the client’s unique business needs.
For more information, questions, or comments, contact us at info@bmtadvisors.com
Follow us on LinkedIn.
Reference:
- Guidance Document Breakthrough Devices Program. Guidance for Industry and Food and Drug Administration Staff. September 15, 2023. https://www.fda.gov/media/162413/download
- Breakthrough Devices Program. https://www.fda.gov/medical-devices/how-study-and-market-your-device/breakthrough-devices-program#authorizations
- Guidance Document. Breakthrough Devices Program. Guidance for Industry and Food and Drug Administration Staff. December 18, 2018. https://www.fda.gov/media/108135/download
- Center for Devices and Radiological Health. 2023 Annual Report. https://www.fda.gov/media/175479/download?attachment
- Safer Technologies Program (SteP) for Medical Devices https://www.fda.gov/medical-devices/how-study-and-market-your-device/safer-technologies-program-step-medical-devices
Photo 272584329 © bpawesome | Dreamstime.com