Advancements in digital health technologies, including artificial intelligence/machine learning-enabled medical devices (MLMD), necessitate developing regulatory…
FDA Expanding Postmarket Surveillance for Medical Devices
The FDA’s Center for Devices and Radiological Health (CDRH) is committed to ensuring the safety and effectiveness of medical devices throughout their lifecycle. A crucial aspect of this oversight is postmarket surveillance, which involves monitoring devices after they enter the market.
Historically, the FDA has relied primarily on passive postmarket surveillance, such as the Medical Device Reporting (MDR) system1. This system depends on voluntary and mandatory reporting of adverse events, such as device-related deaths, serious injuries, and certain device malfunctions, by manufacturers, importers, and device user facilities (e.g., hospitals and nursing homes). Health care professionals, patients, and consumers may also voluntarily submit reports informing FDA about adverse events that have occurred. However, passive surveillance has limitations, including underreporting and delayed reporting of adverse events.

To address the shortcomings of passive surveillance, FDA is increasingly turning to active postmarket surveillance. This approach involves the ongoing analysis of real-world evidence, such as electronic health records and billing claims. By examining these data, the FDA can identify potential safety issues and determine whether regulatory action (e.g., device recall) is necessary.
According to a Government Accountability Office (GAO) investigation, publicly released on Aug 15, 2024, FDA has taken significant steps to establish an active postmarket surveillance system.2 In partnership with the National Evaluation System for Health Technology Coordinating Center (NESTcc), the agency has:
- Organized data contributors: Built a network of healthcare systems and research organizations to provide data. Currently including 19 network collaborators consisting of mostly health systems and research organizations.
- Developed data infrastructure: Created a cloud-based system to collect and analyze real-world evidence.
- Planned surveillance systems: Developed strategies for generating and evaluating evidence to support postmarket surveillance.
FDA plans to begin active surveillance of two specific device types: duodenoscopes and robotically assisted surgical devices used in gallbladder removal (cholecystectomy). These devices were selected based on known safety concerns and the availability of relevant data in electronic health records and claims data. FDA officials estimate this work will be completed by December 2024, contingent on funding availability.
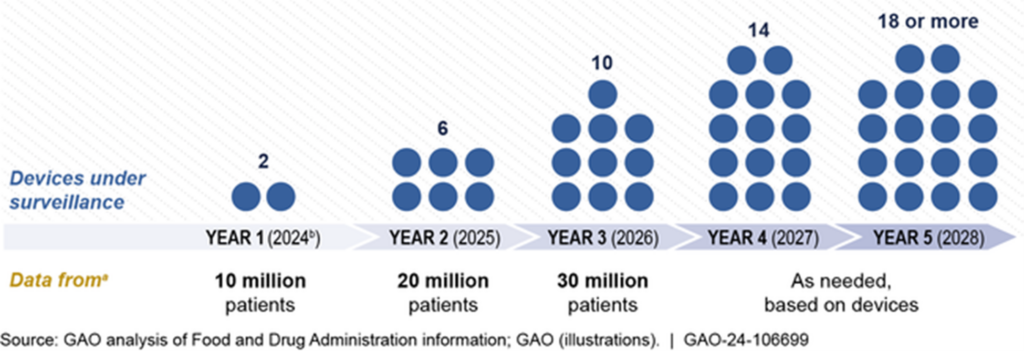
The FDA intends to gradually expand its active surveillance program over the next five years. This expansion will involve adding more device types and onboarding additional data partners to increase the number of patients included in the analysis. Specifically:
- Year 1 (2024): Surveillance of duodenoscopes and robotic surgical devices.
- Years 2 and 3 (2025-2026): Addition of four devices per year and onboarding of data partners to include 10 million new patients each year.
- Subsequent years: Continued expansion as needed.
Planned Expansion of FDA’s Active Postmarket Surveillance:
Despite the progress made, implementing an effective active postmarket surveillance system faces several challenges. These include limited use of unique device identifiers (UDIs)3 and funding constraints. Addressing these challenges will be crucial for ensuring the ongoing safety and effectiveness of medical devices.
As the FDA continues to enhance its postmarket surveillance capabilities, it is essential to balance the need for rigorous oversight with the goal of facilitating innovation and access to life-saving medical technologies. By leveraging both passive and active surveillance approaches, the FDA can better protect public health and ensure that medical devices meet the highest standards of safety and efficacy.
In addition to passive and active postmarket surveillance, the FDA can require manufacturers to conduct postmarket surveillance (i.e., 522 order) at the time of approval or clearance of a medical device, or at any time thereafter if a potential issue is identified.4 This order implies an active, systematic, and scientifically valid collection, analysis, and interpretation of data or other information about the marketed device. Failure to comply with a 522 order (within 15 months of the order) can result in enforcement actions, such as seizure of product, injunction, prosecution, or civil monetary penalties.
The FDA may order postmarket surveillance to address a wide variety of device-related public health questions, including:
- Understanding the nature, severity, or frequency of suspected problems reported in adverse event reports or the published literature.
- Obtaining more information on device performance associated with real-world clinical practice.
- Addressing long-term or infrequent safety and effectiveness issues for devices with limited premarket testing.
- Better defining the association between problems and devices when unexpected or unexplained serious adverse events occur after a device is marketed if there is a change in the nature of serious adverse events (e.g., severity), or if there is an increase in the frequency of serious adverse events.
Data collected through postmarket surveillance may include rates of malfunction or failure of a device intended for long-term use or incidents of latent sequelae resulting from device use. This data helps to address important public health questions on the safety and effectiveness of a device.
There are various types of postmarket surveillance designs, including conducting a prospective or retrospective analysis of data from real-world sources, such as device registries and electronic health records and/or continued monitoring of the distribution and trends in the incidence of adverse events through ongoing, active or passive systematic collection, analysis, and interpretation of data (i.e., MDR-reportable and MDR nonreportable adverse events or device complaints).
The FDA intends to work closely with manufacturers on the development of their surveillance plans, including timelines, expectations, and the posting of interim data on the FDA’s website. The agency has developed the “Postmarket Surveillance Under Section 522 of the Federal Food, Drug, and Cosmetic Act” guidance document4 to facilitate discussions with manufacturers on postmarket surveillance plans, issues, and challenges.
About Boston MedTech Advisors (BMTA):
Since 2004, BMTA’s multidisciplinary team of highly experienced consultants has supported more than 400 medical technologies and life sciences companies around the world to achieve their business goals. BMTA assists its clients to commercialize new products and services and increase their market adoption, by addressing their unique and inter-dependent regulatory, clinical, reimbursement, marketing, and business development requirements. BMTA offers valuable, ethical, result-oriented, professional, and cost-effective insights that recognize the multi-faceted aspects of today’s healthcare markets and the client’s unique business needs.
For more information, questions, or comments, contact us at info@bmtadvisors.com
Follow us on LinkedIn.
References
- Medical Device Reporting (MDR): How to Report Medical Device Problems https://www.fda.gov/medical-devices/medical-device-safety/medical-device-reporting-mdr-how-report-medical-device-problems
- United States Government Accountability Office Report to Congressional Requesters. MEDICAL DEVICES FDA Has Begun Building an Active Postmarket Surveillance System. July 2024. Publicly Released: Aug 15, 2024. https://www.gao.gov/assets/gao-24-106699.pdf
- UDI Basics https://www.fda.gov/medical-devices/unique-device-identification-system-udi-system/udi-basics
- Postmarket Surveillance Under Section 522 of the Federal Food, Drug, and Cosmetic Act. October 7, 2022 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/postmarket-surveillance-under-section-522-federal-food-drug-and-cosmetic-act
Photo 32252864 © Convisum Dreamstime.com