Boston MedTech Advisors Blog, January 2024
In cases where medical devices come in direct or indirect contact with the human body, device manufacturers are required to provide biological evaluation of the device and its material components, in their FDA submissions. This biological evaluation assesses the potential of an unacceptable adverse biological response resulting from contact of the component materials of the device with the body. Biocompatibility (i.e., the ability of a device material to perform with an appropriate response in a specific situation) is the term commonly used to describe the biological requirements of a biomaterial or biomaterials used in a medical device.
In September 2023, FDA released a final guidance document entitled: “Use of International Standard ISO 10993-1, Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process.”
If a device has “no direct or indirect tissue contact components” (e.g., software only devices), all that is required is a confirmatory statement to that effect. However, devices with even transient contact, require an assessment of biocompatibility risk to determine if testing is needed. When biocompatibility testing is warranted, the Agency recommends testing medical devices under conditions of use, whenever possible.
The biological assessment evaluates the final finished device within the framework of a risk management process. The assessment includes the complete medical device, the material components, the manufacturing processes (e.g., sterilization), and the clinical use (e.g., location, frequency, and duration of exposure). Risks may include chemical toxicity, unacceptable biological response to physical characteristics of the device, and aspects of manufacturing and processing that could alter the physicochemical characteristics of the device whereby changing the biocompatibility response. Manufacturers should consider all available information to identify and mitigate risks, including literature and other publicly available information, clinical experience, animal study experience, consensus standards, and devices previously reviewed by FDA.

International Standard ISO 10993-1 “Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process” is recognized by FDA for evaluating the biological safety of medical devices. In September 2023, FDA released a final guidance document entitled: “Use of International Standard ISO 10993-1, Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process”1 to provide clarification and updated information on how to use ISO 10993-1. This guidance supersedes the previous, September 2020 guidance of this standard2 and is based on the draft guidance published October 20203.
The guidance addressed seven main principles of biocompatibility evaluation:
- Selection of materials used in medical devices and its biocompatibility evaluation should initially consider the likelihood of direct or indirect tissue contact, and any available information for the materials of manufacture.
- Materials of manufacture, the device in final form, and possible leachable chemicals or degradation products should be considered.
- Endpoints relevant to biocompatibility evaluation should consider the nature, degree, frequency, duration, and conditions of exposure of the device materials to the body.
- All in vitro or in vivo bio safety experiments conducted in accordance with Good Laboratory Practice (GLP) regulation.
- When test data are provided, complete experimental data should be submitted.
- Any change in chemical composition, manufacturing process, physical configuration, or intended use should be evaluated with respect to possible changes in biocompatibility and need for additional biocompatibility testing.
- Evaluation performed according to the ISO standard should be considered in conjunction with information obtained from other nonclinical tests, clinical studies, and post market experiences.
The new 2023 guidance represents FDA’s current thoughts on the use of ISO 10993-1 in the evaluation of the biological safety of medical devices. While few major changes were made from the 2020 guidance, the following changes are important to note when implementing this standard for medical device submissions:
These major changes presented herein, in addition to other minor revisions in the new guidance compared to the 2020 version, may reduce the burden of biological safety documentation required when submitting a premarket application.
- Certain devices in contact with intact skin, that are fabricated from common polymers and fabrics, no longer require biocompatibility testing.
- These devices pose a very low biocompatibility risk due to their long history of safe use in legally marketed medical devices. Based on Quality System Regulation and other post market controls to identify potential biocompatibility related issues, premarket submission would only require specific material information in these cases.
- The guidance specifies types of medical devices and specific materials that are included in this policy. The list of device materials and exclusion characteristics will be periodically reassessed by FDA.
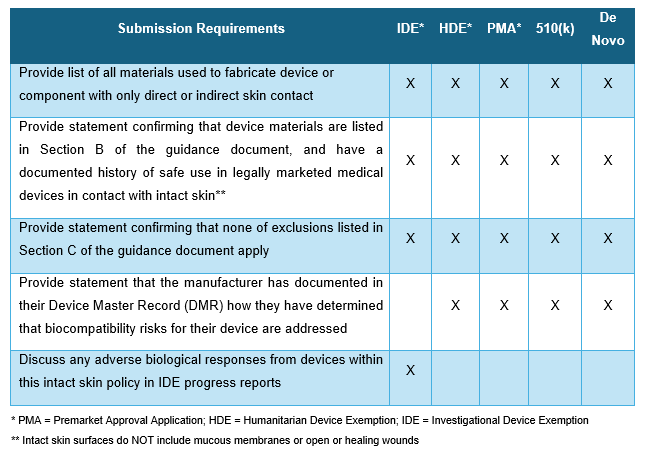
- The biocompatibility information needed per submission type listed in the table below.
- eSTAR, an interactive PDF form that guides applicants through the process of preparing a comprehensive medical device submission, has changed to accommodate the changes in this guidance document.
- Based on this guidance, intact skin contacting components may include, for example:
- The head/chin rest of the biomicroscope used in the ophthalmologist office for microscopic assessments of the eye.
- Finger cushion and interactive components of the oximeter device that slides over the fingertip to measure blood oxygen levels.
- A blood pressure cuff wrapped around the arm.
- Leg wraps of powered inflatable tube massager device that uses air compression to help relieve minor muscle aches and increase leg circulation.
- FDA feedback, via pre-submission, is recommended in certain cases, for example:
- If a legally marketed device made from same material resulted in adverse biocompatibility-related findings after marketing.
- The device is indicated for use with neonates or in pregnant women.
- The device is a combination product, or a device comprised of biologically-derived material.
- Inclusion of language about Accreditation Scheme for Conformity Assessment (ASCA) Program.4 The ASCA is a voluntary accreditation scheme intended to:
- Streamline conformity assessment in device submissions
- Enhance FDA’s confidence in methods and results
- Decrease the need for additional information related to conformance with a standard
- Promote consistency, predictability, and efficiency in medical device review
- Serve as a least burdensome approach.
- Under this program, testing laboratories may be accredited to perform premarket testing for device manufacturers for premarket submissions. In the new guidance, FDA states that an ASCA Summary Test Report is sufficient for testing conducted under this program, rather than a complete study report. Thus, the use of ASCA accredited labs may reduce the documentation required to be submitted to FDA.
These major changes presented herein, in addition to other minor revisions in the new guidance compared to the 2020 version, may reduce the burden of biological safety documentation required when submitting a premarket application. Device manufacturers should keep abreast of periodical re-evaluation of materials and consequent changes to the material list published by FDA. The guidance’s detailed and specific instructions on the process of risk assessment, specific tests, and submission requirements should be considered early in the design process.
About Boston MedTech Advisors (BMTA):
Since 2004, BMTA’s multidisciplinary team of highly experienced consultants has supported more than 400 medical technologies and life sciences companies around the world to achieve their business goals. BMTA assists its clients to commercialize new products and services and increase their market adoption, by addressing their unique and inter-dependent regulatory, clinical, reimbursement, marketing, and business development requirements. BMTA offers valuable, ethical, result-oriented, professional, and cost-effective insights that recognize the multi-faceted aspects of today’s healthcare markets and the client’s unique business needs.
For more information, questions, or comments, contact us at info@bmtadvisors.com
Follow us on LinkedIn.
References:
- Guidance for Industry and Food and Drug Administration Staff “Use of International Standard ISO 10993-1, ‘Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process.’” September 2023. https://www.fda.gov/media/142959/download
- Guidance for Industry and Food and Drug Administration Staff “Use of International Standard ISO 10993-1, ‘Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process.’” September 4, 2020. https://www.fda.gov/media/85865/download
- Draft Guidance for Industry and Food and Drug Administration Staff “Select Updates for Biocompatibility of Certain Devices in Contact With Intact Skin.” October 15, 2020. https://www.federalregister.gov/documents/2020/10/15/2020-22876/select-updates-for-biocompatibility-of-certain-devices-in-contact-with-intact-skin-draft-guidance
- Accreditation Scheme for Conformity Assessment (ASCA) https://www.fda.gov/medical-devices/standards-and-conformity-assessment-program/accreditation-scheme-conformity-assessment-asca
Photo 120283606 © Dmitry Kotin | Dreamstime.com